|
|
|
(Allegato V) - Al giorno d'oggi, per quanto rigurda l'analisi degli oli vegetali, è indispensabile l'analisi della frazione sterolica. I motivi sono da ricercarsi nel fatto che la frazione sterolica non è influenzabile da variazioni genetiche che invece possono mutare radicalmente la componente saponificabile e quella degli acidi grassi. |
|
|
1. OGGETTO 2. PRINCIPIO DEL METODO 3.1. Matraccio da 250 ml, munito di refrigerante a ricadere con giunti a smeriglio. 3.2. Imbuti separatori da 500 ml. 3.3. Matracci da 250 ml. 3.4. Attrezzatura completa per analisi cromatografica su strato sottile, per lastre di vetro 20 * 20 cm. 3.5. Lampada a luce ultravioletta, con lunghezza d'onda 366 o 254 nm. 3.6. Microsiringhe da 100 µl e 500 µl. 3.7. Imbuto cilindrico filtrante a setto poroso G 3 (porosità 15-40 µm) di diametro circa 2 cm e altezza circa 5 cm, con attacco idoneo per filtrazione sotto vuoto e giunto smerigliato maschio 12/21. 3.8. Beuta per vuoto da 50 ml con giunto femmina smerigliato 12/21 adattabile all'imbuto filtrante (3.7.). |
Figura 1 - Gascromatogramma
della frazione sterolica di un olio di oliva grezzo |
| 3.9.
Provetta da 10 ml a fondo conico con tappo a tenuta. 3.10. Gascromatografo idoneo per il funzionamento con colonna capillare, dotato di sistema di splittaggio, costituito da: 3.10.1. Camera termostatica per le colonne, idonea a mantenere la temperatura desiderata con la precisione di ± 1°C. 3.10.2. Complesso di vaporizzazione termoregolabile con elemento vaporizzante in vetro persilanizzato. 3.10.3. Rivelatore a ionizzazione di fiamma e convertitore-amplificatore. 3.10.4. Registratore-integratore idoneo per il funzionamento con il convertitore-amplificatore (3.10.3.), con tempo di risposta non superiore a 1 secondo e con velocità della carta variabile. 3.11. Colonna capillare in vetro o silice fusa, lunga 20 + 30 m, diametro interno 0,25 + 0,32 mm, internamente ricoperta con liquido SE-52 o SE-54 o equivalenti, con spessore uniforme compreso fra 0,10 e 0,30 µm. 3.12. Microsiringa per gascromatografia da 10 µl con ago cementato .4. REAGENTI 4.1. Potassio idrossido, soluzione etanolica circa 2 N: si sciolgono, sotto raffreddamento, 130 g di idrossido di potassio (titolo minimo 85 %) in 200 ml di acqua distillata, quindi si porta ad 1 litro con etanolo. La soluzione si conserva in bottiglie di vetro scuro ben tappate. 4.2. Etere etilico, puro per analisi. 4.3. Sodio solfato anidro, puro per analisi. 4.4. Lastre di vetro stratificate con gel di silice, senza indicatore di fluorescenza, spessore 0,25 mm (sono reperibili in commercio già pronte per l'uso). |
Figura
2 - Gascromatogramma della frazione sterolica di un olio di
oliva raffinato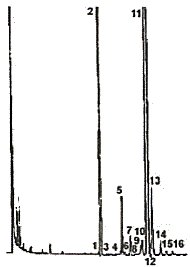 |
| 4.5. Potassio
idrossido, soluzione etanolica 0,2 N: si sciolgono 13 g di idrossido di
potassio in 20 ml di acqua distillata e si porta a 1 litro con etanolo. 4.6. Benzene, per cromatografia (vedi 5.2.2). 4.7. Acetone, per cromatografia (vedi 5.2.2). 4.8. Esano, per cromatografia (vedi 5.2.2). 4.9. Etere etilico, per cromatografia. 4.10. Cloroformio, puro per analisi. 4.11. Soluzione di riferimento per la cromatografia su placca: colesterolo o fitosteroli, soluzione al 5 % in cloroformio. 4.12. 2,7-Diclorofluoresceina, soluzione etanolica allo 0,2 %. Si rende leggermente basica aggiungendo qualche goccia di soluzione alcolica 2 N di idrossido di potassio. 4.13. Piridina anidra, per cromatografia. 4.14. Esametildisilazano. 4.15. Trimetilclorosilano. 4.16. Soluzioni campione di trimetilsilileteri degli steroli: si preparano al momento dell'impiego partendo da steroli puri o da miscele di steroli ottenute da oli che li contengano. 4.17. a-colestanolo, soluzione allo 0,2 % (m/V) in cloroformio (standard interno). 4.18. Gas vettore: idrogeno o elio, puri per gascromatografia. 4.19. Gas ausiliari: - idrogeno, puro per gascromatografia - aria, pura per gascromatografia. |
|
|
|
|
| 5. PROCEDIMENTO 5.1. Preparazione dell'insaponificabile. 5.1.1. Nel matraccio da 250 ml si introduce, impiegano la microsiringa da 500 µl, un volume di soluzione di a -colestanolo allo 0,2 % in cloroformio (4.17.) che contenga una quantità di a -colestanolo corrispondente a circa il 10 % del contenuto di steroli nell'aliquota di campione da prelevare per la determinazione. Ad esempio per 5 g di campione si aggiungano 500 µl della soluzione di a-colestanolo allo 0,2 % se trattasi di un olio di oliva e 1 500 µl se trattasi di oli di semi o olio di sansa di oliva. Si evapora in corrente di azoto fino a secchezza, quindi nello stesso matraccio si pesano esattamente 5 g di campione secco e filtrato. In caso di oli e grassi animali o vegetali contenenti quantità notevoli di colesterolo può essere presente un picco avente tempo di ritenzione identico al colestanolo. In tali casi occorre analizzare la frazione sterolica in doppio con e senza standard interno. 5.1.2. Si aggiungono 50 ml di soluzione etanolica di idrossido di potassio 2 N, si applica il refrigerante a ricadere e si scalda a leggera ebollizione su bagnomaria sotto continua energica agitazione, fino a saponificazione avvenuta (la soluzione diviene limpida). Si continua il riscaldamento ancora per 20 minuti, quindi si aggiungono 50 ml di acqua distillata facendoli scendere dall'alto del refrigerante, si stacca il refrigerante e si raffredda il matraccio a circa 30°C. 5.1.3. Si travasa il contenuto del matraccio quantitativamente, in un imbuto separatore da 500 ml, aiutandosi con acqua distillata, a più riprese, impiegandone complessivamente circa 50 ml. Si aggiungono circa 80 ml di etere etilico, si agita energicamente per circa 30 secondi e si lascia stratificare (nota 1). Si separa la fase acquosa sottostante raccogliendola in un secondo imbuto separatore. Sulla fase acquosa si effettuano ancora due estrazioni, con le stesse modalità, impiegando ogni volta 60-70 ml di etere etilico. Nota 1 - Eventuali emulsioni possono essere eliminate aggiungendo, mediante spruzzetta, piccole quantità di alcool etilico o metilico. 5.1.4. Si riuniscono gli estratti eterei in un unico imbuto separatore e si lavano con acqua distillata (50 ml per volta) fino a reazione neutra delle acque di lavaggio. Eliminata l'acqua di lavaggio, si essicca con solfato di sodio anidro e si filtra, su solfato sodico anidro, in un matraccio da 250 ml previamente pesato, lavando imbuto e filtro con piccole quantità di etere etilico. 5.1.5. Si distilla l'etere fino a pochi ml, quindi si porta a secco sotto leggero vuoto o in corrente di azoto, si completa l'essiccamento in stufa a 100°C per un quarto d'ora circa e, dopo raffreddamento in essiccatore, si pesa. 5.2. Separazione della frazione sterolica. 5.2.1. Preparazione delle lastre basiche: si immergono le lastre al gel di silice (4.4.), completamente, nella soluzione etanolica 0,2 N di idrossido di potassio (4.5.) per 10 secondi, si lasciano quindi asciugare sotto cappa per 2 ore ed infine si pongono in stufa a 100°C per 1 ora. Si tolgono dalla stufa e si conservano in essiccatore a cloruro di calcio fino al momento dell'impiego (le placche così trattate devono essere impiegate entro 15 giorni). Nota 2 - Impiegando per la separazione della frazione sterolica delle lastre di gel di silice basiche si elimina la necessità del trattamento dell'insaponificabile con allumina. In tal modo vengono trattenuti sulla linea di caricamento tutti i composti di natura acida (acidi grassi ed altro) ottenendosi così la banda degli steroli nettamente separata dalle bande degli alcoli alifatici e triterpenici. 5.2.2. Nella camera di sviluppo delle lastre si introduce una miscela benzene-acetone 95:5 (V/V) fino all'altezza di circa 1 cm. In alternativa può essere usata una miscela esano-etere etilico 65:35 (V/V). Si chiude la camera con l'apposito coperchio e si lascia così per almeno mezz'ora in modo che si stabilisca l'equilibrio liquido-vapore. Sulle superfici interne della camera possono essere fissate delle strisce di carta da filtro che peschino nell'eluente: questo accorgimento permette di ridurre di circa 1/3 il tempo di sviluppo e di ottenere una più uniforme e regolare eluizione dei componenti. Nota 3 - Al fine di ottenere condizioni di eluizione perfettamente riproducibili la miscela di sviluppo deve essere sostituita ad ogni prova. 5.2.3. Si prepara una soluzione al 5 % circa di insaponificabile (5.1.5.) in cloroformio e, con la microsiringa da 100 µl si depositano su una placca cromatografica (5.2.1.) a 2 cm circa da una estremità, 0,3 ml di detta soluzione, in striscia il più possibile sottile ed uniforme. In allineamento con la linea di caricamento, ad un'estremità della lastra si depositano 2-3 µl della soluzione di riferimento degli steroli (4.11.), allo scopo di identificare, a sviluppo ultimato, la banda degli steroli. 5.2.4. Si pone la placca nella camera di sviluppo preparata come detto in 5.2.2. La temperatura ambiente dovrà essere mantenuta fra 15 e 20°C. Si chiude subito la camera col coperchio e si lascia eluire fino a che il fronte del solvente sia arrivato a circa 1 cm dal bordo superiore della placca. Si rimuove quindi la placca dalla camera di sviluppo e si evapora il solvente in corrente di aria calda oppure lasciando la placca per un pò di tempo sotto cappa. 5.2.5. Si spruzza la placca debolmente ed uniformemente con la soluzione di 2,7-diclorofluoresceina. Osservando la lastra alla luce ultravioletta si individua la banda degli steroli per allineamento con la macchia ottenuta con la soluzione di riferimento; si delimitano con una matita nera i limiti della banda lungo i margini di fluorescenza. 5.2.6. Con una spatola metallica si raschia il gel di silice compreso nell'area delimitata. Il materiale asportato, finemente sminuzzato, viene introdotto nell'imbuto filtrante (3.7.); si aggiungono 10 ml di cloroformio caldo, si mescola accuratamente con la spatola metallica e si filtra aiutandosi con il vuoto, raccogliendo il filtrato nella beuta (3.8.) collegata all'imbuto filtrante. Si lava il residuo nell'imbuto per tre volte con etere etilico (circa 10 ml per volta) raccogliendo sempre il filtrato nella stessa beuta adattata all'imbuto. Si evapora il filtrato fino ad un volume di circa 4-5 ml, si trasferisce la soluzione residua nella provetta da 10 ml (3.9.) previamente pesata, si porta a secco con blando riscaldamento in leggera corrente di azoto, si riprende con qualche goccia di acetone, si riporta ancora a secco, si pone 10 minuti circa in stufa a 105°C indi si lascia raffreddare in essiccatore e si pesa. Il residuo contenuto nella provetta è costituito dalla frazione sterolica. 5.3. Preparazione dei trimetilsilileteri. 5.3.1. Nella provetta contenente la frazione sterolica si aggiunge il reattivo per la sililazione, costituito da una miscela di piridina-esametildisilazano-trimetilclorosilano 9:3:1 (V/V/V) (nota 4) in ragione di 50 µl per ogni milligrammo di steroli, evitando ogni assorbimento di umidità (nota 5). Nota 4 - Esistono in commercio soluzioni già pronte per l'uso; sono inoltre disponibili altri reagenti silanizzanti, quali ad esempio il bis-trimetiltrifluorolacetammide + 1 % trimetilclorosilano da diluire son uno stesso volume di piridina anidra. 5.3.2. Si tappa la provetta, si agita cautamente (senza capovolgere) fino a completa solubilizzazione degli steroli. Si lascia a sé per almeno 15 minuti a temperatura ambiente, quindi si centrifuga per alcuni minuti: la soluzione limpida è pronta per l'analisi gascromatografica. Nota 5 - L'eventuale formazione di una leggera opalescenza è normale e non è causa di alcun disturbo. La formazione di un flocculato bianco o la comparsa di una colorazione rosa sono indizio della presenza di umidità o di alterazione del reattivo. In questo caso la prova dovrà essere ripetuta. 5.4. Analisi gascromatografica. 5.4.1. Operazioni preliminari, condizionamento della colonna. 5.4.1.1. Si installa nel gascromatografo la colonna, collegando il terminale di ingresso all'evaporatore connesso col sistema di splittaggio e il terminale di uscita al rivelatore. Si eseguono i controlli generali del complesso gascromatografico (tenuta dei circuiti dei gas, efficienza del rivelatore, efficienza del sistema di splittaggio e del sistema di registrazione, ecc.). 5.4.1.2. Se la colonna è messa in uso per la prima volta è consigliabile procedere al suo condizionamento. Si fa fluire un leggero flusso di gas attraverso la colonna stessa, quindi si accende il complesso gascromatografico e si inizia un riscaldamento graduale fino a raggiungere una temperatura di almeno 20°C superiore a quella di esercizio (nota 6). Si mantiene tale temperatura per almeno 2 ore, quindi si porta il complesso alle condizioni di funzionamento (regolazione del flusso dei gas e dello splittaggio, accensione della fiamma, collegamento con il registratore elettronico, regolazione della temperatura della camera per la colonna, del rivelatore e dell'iniettore, ecc.) e si registra il segnale ad una sensibilità almeno 2 volte superiore a quella prevista per l'esecuzione dell'analisi. Il tracciato della linea di base deve risultare lineare, esente da picchi di qualsiasi natura, e non deve presentare deriva. Una deriva rettilinea negativa indica imperfetta tenuta delle connessioni della colonna, una deriva positiva indica un insufficiente condizionamento della colonna. Nota 6 - La temperatura di condizionamento deve in ogni caso essere inferiore di almeno 20°C alla temperatura massima prevista per il liquido di ripartizione impiegato. 5.4.2. Scelta delle condizioni operative. 5.4.2.1. Condizioni operative di massima sono le seguenti: - temperatura della colonna: 260°C ± 5°C - temperatura dell'evaporatore: 280°C - temperatura del rivelatore: 290°C - velocità lineare del gas di trasporto: elio 20 + 35 cm/s, idrogeno 30 + 50 cm/s - rapporto di splittaggio: da 1:50 a 1:100 - sensibilità strumentale: da 4 a 16 volte l'attenuazione minima - sensibilità di registrazione: 1 + 2 mV f.s. - velocità della carta: 30 + 60 cm/ora - quantità di sostanza iniettata: 0,5 + 1 µl di soluzione di TMSE. Tali condizioni possono essere modificate in funzione delle caratteristiche della colonna e del gascromatografo in modo da ottenere cromatogrammi che soddisfino le condizioni seguenti: - il tempo di ritenzione del b -sitosterolo deve essere 20 ± 5 minuti - il picco del campesterolo deve essere: per l'olio di oliva (contenuto medio 3 %) 15 ± 5 % del fondo scala, per l'olio di soia (contenuto medio 20 %) 80 ± 10 % del fondo scala - si deve avere separazione di tutti gli steroli presenti; è necessario che i picchi oltre che separati siano anche completamente risolti cioè che il tracciato del picco raggiunga la linea di base prima dell'uscita del picco successivo. È tuttavia tollerata anche una risoluzione incompleta a condizione però che sia quantificabile secondo la perpendicolare il picco a TRR 1,02. 5.4.3. Esecuzione dell'analisi. 5.4.3.1. Con la microsiringa da 10 µl si preleva 1 ml di esano, si aspirano 0,5 µl di aria e successivamente 0,5 + 1 µl della soluzione del campione; si alza ancora lo stantuffo della siringa in modo che l'ago sia vuoto. Si introduce l'ago attraverso la membrana del complesso di iniezione e dopo 1-2 secondi si inietta rapidamente e si estrae quindi lentamente l'ago dopo circa 5 secondi. 5.4.3.2. Si effettua la registrazione fino a completa eluizione dei TMSE degli steroli presenti. La linea di base deve essere sempre corrispondente ai requisiti richiesti (5.4.1.2.). 5.4.4. Identificazione dei picchi. L'identificazione dei singoli picchi viene effettuata in base ai tempi di ritenzione e per paragone con miscele di TMSE degli steroli, analizzate nelle medesime condizioni. Gli steroli vengono eluiti secondo il seguente ordine: colesterolo, brassicasterolo, 24-metilencolesterolo, campesterolo, campestanolo, stigmasterolo, D 7-campesterolo, D 5'23-stigmastadienolo, clerosterolo, b -sitosterolo, sitostanolo, D 5-avenasterolo, D 5'24-stigmastadienolo, D 7-stigmastenolo, D 7-avenasterolo. Nella Tabella I sono riportati i tempi di ritenzione relativi al sitosterolo per le colonne SE 52 e SE 54. Le figure 1 e 2 illustrano cromatogrammi tipici di alcuni oli. 5.4.5. Valutazione quantitativa. 5.4.5.1. Si procede al calcolo con l'integratore, delle aree dei picchi dell'a-colestanolo e degli steroli. Non vengono considerati i picchi di eventuali componenti non compresi fra quelli elencati nella Tabella I. Il coefficiente di risposta dell'a-colestanolo si deve intendere unitario. 5.4.5.2. Si calcola il contenuto di ogni singolo sterolo, in mg/100 g di sostanza grassa, come segue: |
|
| sterolo x = (AX * ms * 100 / As * m) | |
|
in cui: 6. ESPRESSIONE DEI RISULTATI |
|
| % Dello sterolo x = (AX / å A) * 100 | |
| in cui: AX = Area del picco X, å A = Sommatoria delle aree di tutti i picchi. APPENDICE Determinazione della velocità lineare dei gas. Nel gascromatografo, regolato alle normali condizioni operative, si iniettano 1 + 3 ml di metano (o propano) e si cronometra il tempo che il gas impiega a percorrere la colonna, dal momento dell'iniezione al momento dell'uscita del picco (tM). La velocità lineare in cm/s è data da L/tM in cui L è la lunghezza della colonna in centimetri e tM è il tempo cronometrato in secondi |
|
| . |
|
|
Tabella 1
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figura 1 - Gascromatogramma della frazione sterolica di un olio di oliva grezzo
|
Figura 2 - Gascromatogramma della frazione sterolica di un olio di oliva raffinato
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 analisi steroli |
 analisi steroli |
|
 |
esame della lastra TLC alla luce UV per evidenziare la banda degli steroli |